
文章肯定了ctDNA水平变化作为药物临床试验的早期终点的潜力和可行性,在介绍了影响ctDNA作为临床试验早期临床替代终点的各类因素的基础上,呼吁各方在今后的临床试验开展中采用统一的规范积极推进ctDNA水平变化作为药物临床试验的早期终点的进程。

文章内容主要包括四个部分:
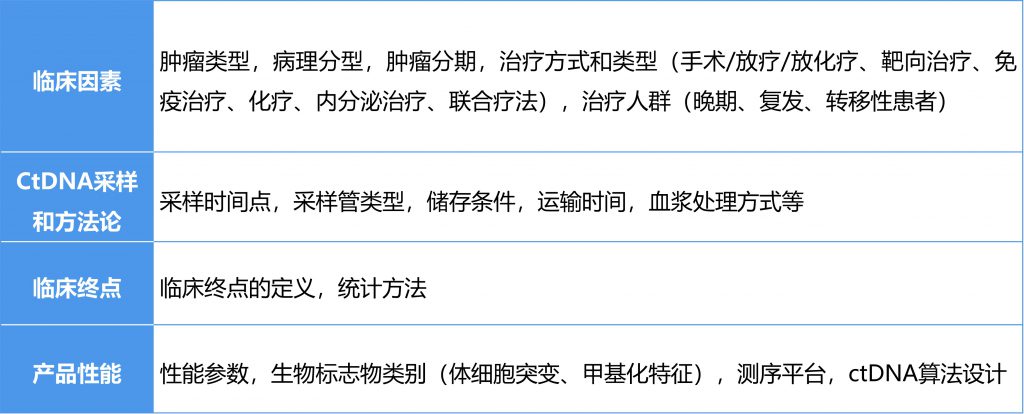
- 支持使用ctDNA水平变化作为早期终点需要考虑的临床问题
- 验证使用ctDNA作为早期终点需要考虑的关键技术/临床问题
- 知识不足及应对方案
- 肿瘤药物开发中使用ctDNA在法规层面的考量
美国FDA今年发布了如何在早期实体瘤药物开发中使用ctDNA的指南草案(点击查看原文),欧洲ESMO也发布了相关的指导原则(点击查看原文),ctDNA加速抗肿瘤药物开发获得业界越来越多的认可,FoCR本次发布的文章也明确指出:FDA支持研究和理解ctDNA成为肿瘤治疗的早期终点,不过需要更多的临床试验数据和meta分析来支持ctDNA成为早期临床替代终点。
目前越来越多的研究证实ctDNA水平降低、MRD清除与患者的预后改善关系密切,但单独研究并不足以支持使用ctDNA作为经验证的替代终点。而且不同临床试验、不同适应症、不同检测方法以及不同地域/人种的基因数据为这些研究的数据整合带来了极大的困难,不利于快速支持ctDNA成为临床替代终点。
慧渡医疗新一代个体化MRD检测产品已获得CE认证(点击查看原文)。该MRD检测产品是一款不依赖肿瘤组织、可利用液态活检样本实现的定制化MRD检测产品。通过血液或尿液样本进行外显子组测序鉴别患者个性化的基因突变,挑选多至50个患者特异的体细胞突变、结合500个临床用药相关突变位点共同设计个体化MRD panel,从而实现极低频(0.005%)基因突变检测,进行患者疗效监控,具有重要的临床应用价值。(点击查看原文)。
慧渡医疗自主开发的PredicineATLAS™ cfDNA 600产品在知名药企百时美施贵宝(BMS)对5款国际认可度高的大panel液态活检产品开展的性能测试比较中脱颖而出(点击查看原文)。该检测产品在AstraZeneca开展的一项晚期胆道肿瘤患者免疫治疗的II期临床研究中,证实了血浆ctDNA水平下降与疗效相关(点击查看原文)。
渡医疗多款基因检测产品获得欧盟CE认证和英国UKCA认证(点击查看原文), 最近获得美国FDA突破性医疗器械(BDD)认定(点击查看原文),这是国际权威监管机构对慧渡医疗技术、产品和其临床应用价值的认可。
慧渡医疗坚持 “起点就是业界顶点” 的从业标准,打造液态活检行业精品,通过在中美同步运营的CAP和CLIA实验室网络,提供中美一致的检测产品和服务,为药企临床试验和患者提供全球一致的检测服务和一站式生物标志物分析。为ctDNA早日成为被认可的临床替代终点提供坚实的临床研究证据,为早日实现肿瘤患者实现精准医疗个性化治疗贡献力量。